
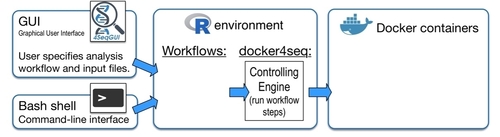
The aim of Reproducible Bioinformatics project is the creation of easy to use Bioinformatics workflows that fullfill the following roles (Sandve et al. PLoS Comp Biol. 2013):
- For Every Result, Keep Track of How It Was Produced
- Avoid Manual Data Manipulation Steps
- Archive the Exact Versions of All External Programs Used
- Version Control All Custom Scripts
- Record All Intermediate Results, When Possible in Standardized Formats
- For Analyses That Include Randomness, Note Underlying Random Seeds
- Always Store Raw Data behind Plots
- Generate Hierarchical Analysis Output, Allowing Layers of Increasing Detail to Be Inspected
- Connect Textual Statements to Underlying Results
- Provide Public Access to Scripts, Runs, and Results
The paper on the SeqBox project is on Bioinformatics (Beccuti et al. 2018).
The paper on the Reproducible Bioinformatics project is on BMC Bioinformatics (Kulkarni et al. 2018).
The paper on rCASC: reproducible classification analysis of single-cell sequencing data is on GigaScience (Alessandri et al. 2019)
The registration page for 7th Edition of RNAseq and Single Cell RNseq workshop is now available also on Elixir training platform